library(iSEE)
library(scRNAseq)
library(scater)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
# launch the app itself ----
app <- iSEE(sce, initial = list(
ColumnDataPlot(
PanelWidth = 8L,
YAxis = "NREADS",
XAxis = "Column data",
XAxisColumnData = "driver_1_s",
ColorBy = "Column data",
ColorByColumnData = "driver_1_s",
FacetColumnBy = "Column data",
FacetColumnByColData = "Core.Type"
)
))
if (interactive()) {
shiny::runApp(app, port=1234)
}Panels
This page showcases panels implemented in iSEE and its known extensions.
Panels are grouped by package in which their are implemented (see the floating table of contents on the right).
Each panel is introduced by a brief description above a single screenshot that illustrates a representative output, and the code used to produce that particular panel output in a live app.
Note
Bear in mind that all those panel classes come with many options to alter their respective outputs. This gallery showcases only a fraction of what each of those panels can do. In all likelihood, if a panel seems to do almost what you have in mind, then there are options to make it do exactly that. Otherwise, options can be added, and more panel classes can be created; check out our resources to learn how!
iSEE
ColumnDataPlot
Visualise any combination of sample metadata stored in a SummarizedExperiment object.
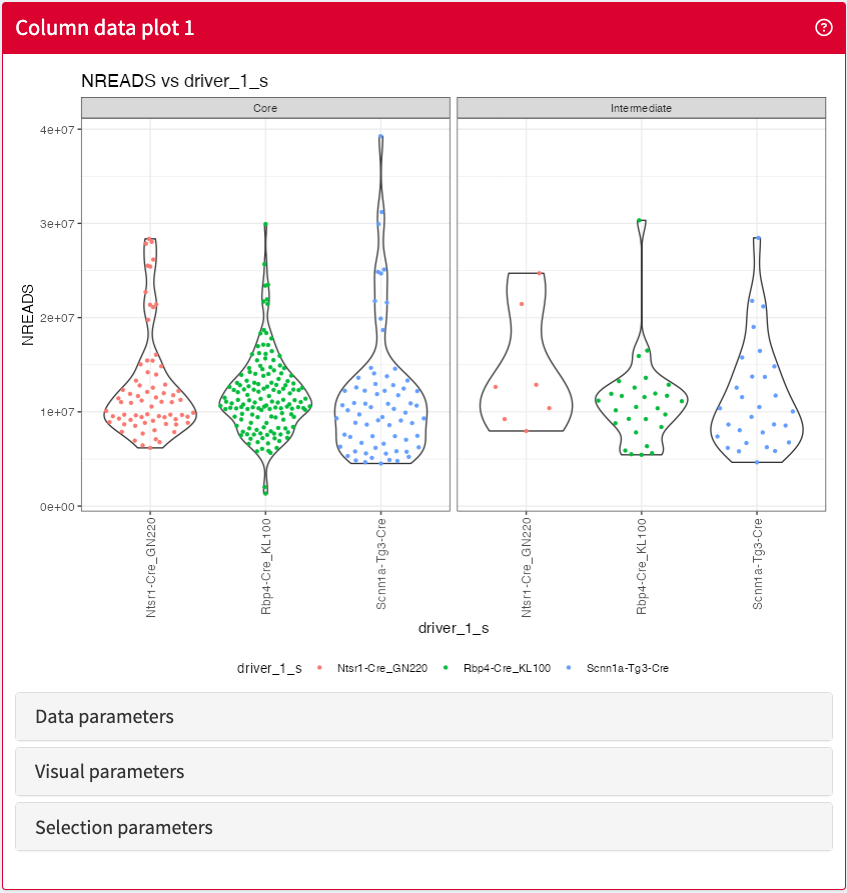
ColumnDataPlot panel class.
Reproduce This Output
ColumnDataTable
Browser and filter sample metadata stored in a SummarizedExperiment object.
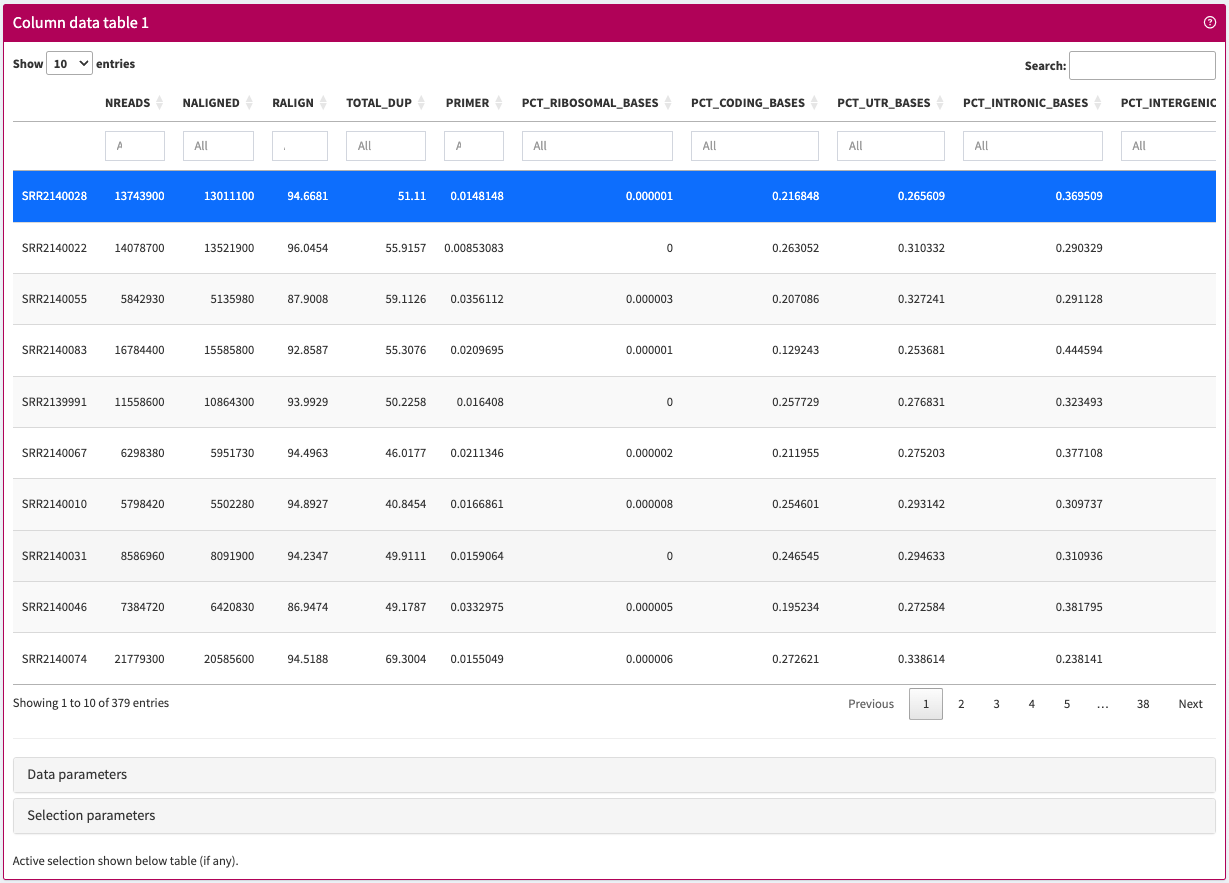
ColumnDataTable panel class.
Reproduce This Output
library(iSEE)
library(scRNAseq)
library(scater)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
# launch the app itself ----
app <- iSEE(sce, initial = list(
ColumnDataTable(
PanelWidth = 12L
)
))
if (interactive()) {
shiny::runApp(app, port=1234)
}ComplexHeatmapPlot
Visualise any number of features and samples in any assay stored in a SummarizedExperiment object.
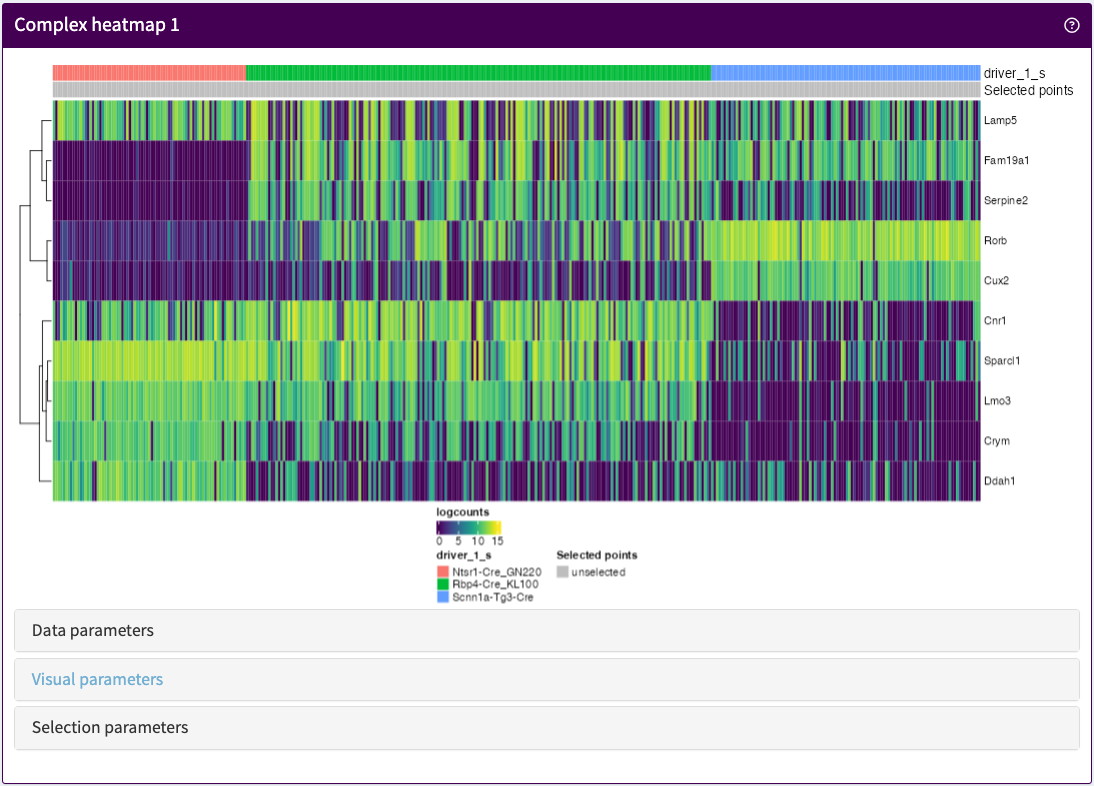
ComplexHeatmapPlot panel class.
Reproduce This Output
library(iSEE)
library(scRNAseq)
library(scater)
library(tibble)
library(dplyr)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
rowData(sce)$ave_count <- rowMeans(assay(sce, "tophat_counts"))
rowData(sce)$n_cells <- rowSums(assay(sce, "tophat_counts") > 0)
rowData(sce)$row_var <- rowVars(assay(sce, "logcounts"))
# launch the app itself ----
# top 10 genes with highest variance in logcounts
gene_list <- c("Lamp5", "Fam19a1", "Cnr1", "Rorb", "Sparcl1", "Crym", "Lmo3", "Serpine2", "Ddah1", "Cux2")
app <- iSEE(sce, initial = list(
ComplexHeatmapPlot(
PanelWidth = 12L,
CustomRows = TRUE,
CustomRowsText = paste0(paste0(gene_list, collapse = "\n"), "\n"),
ColumnData = "driver_1_s",
RowData = "row_var"
)
))
if (interactive()) {
shiny::runApp(app, port=1234)
}FeatureAssayPlot
Visualise up to two features in any assay stored in a SummarizedExperiment object.
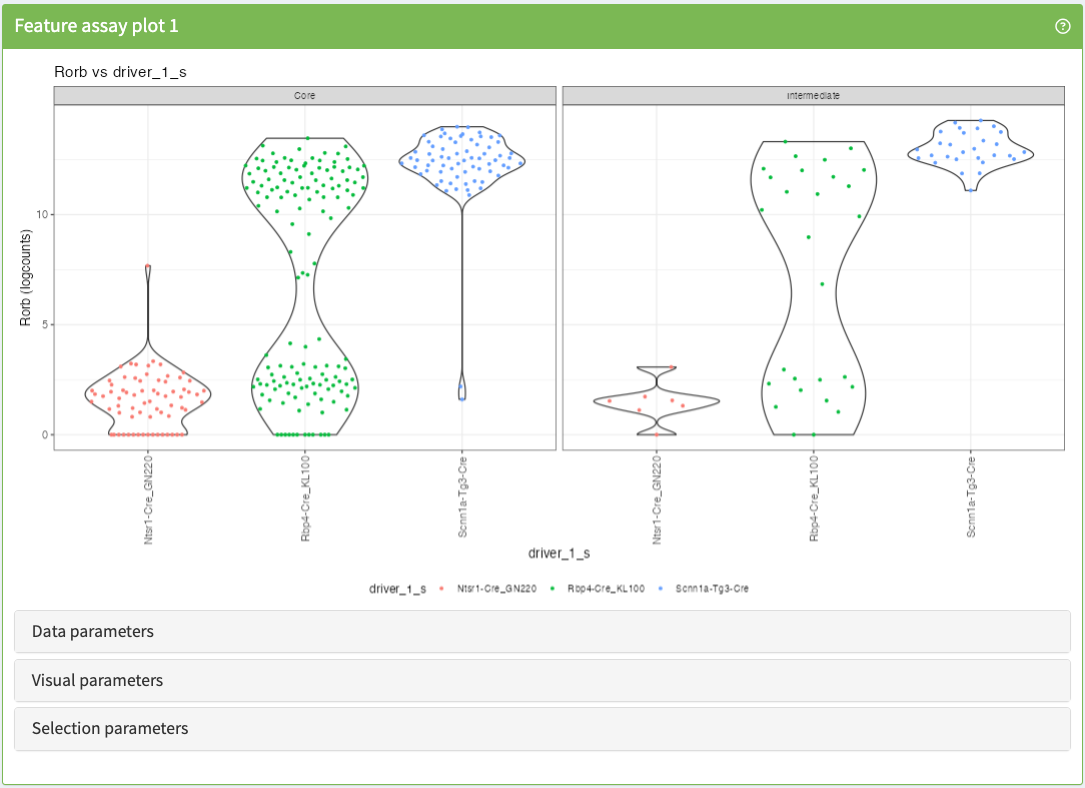
FeatureAssayPlot panel class.
Reproduce This Output
library(iSEE)
library(scRNAseq)
library(scater)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
# launch the app itself ----
app <- iSEE(sce, initial = list(
FeatureAssayPlot(
PanelWidth = 12L,
YAxisFeatureName = "Rorb",
XAxis = "Column data", XAxisColumnData = "driver_1_s",
ColorBy = "Column data", ColorByColumnData = "driver_1_s",
FacetColumnBy = "Column data", FacetColumnByColData = "Core.Type"
)
))
if (interactive()) {
shiny::runApp(app, port=1234)
}ReducedDimensionPlot
Visualise any two dimensions of any dimensionality reduction result stored in a SingleCellExperiment object.
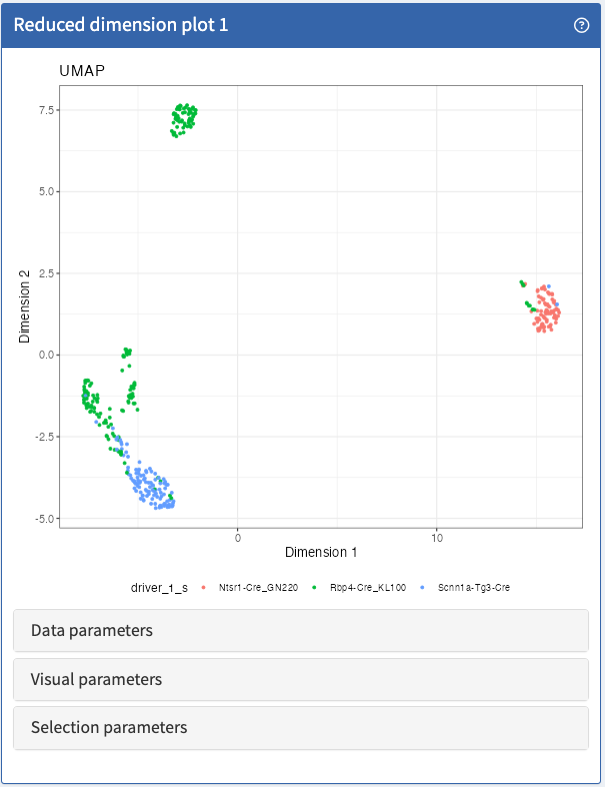
ReducedDimensionPlot panel class.
Reproduce This Output
library(iSEE)
library(scRNAseq)
library(scater)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
sce <- runPCA(sce, ncomponents=4)
sce <- runUMAP(sce)
# launch the app itself ----
app <- iSEE(sce, initial = list(
ReducedDimensionPlot(
PanelWidth = 8L,
Type = "UMAP",
ColorBy = "Column data", ColorByColumnData = "driver_1_s")))
if (interactive()) {
shiny::runApp(app, port=1234)
}RowDataPlot
Visualise any combination of feature metadata stored in a SummarizedExperiment object.
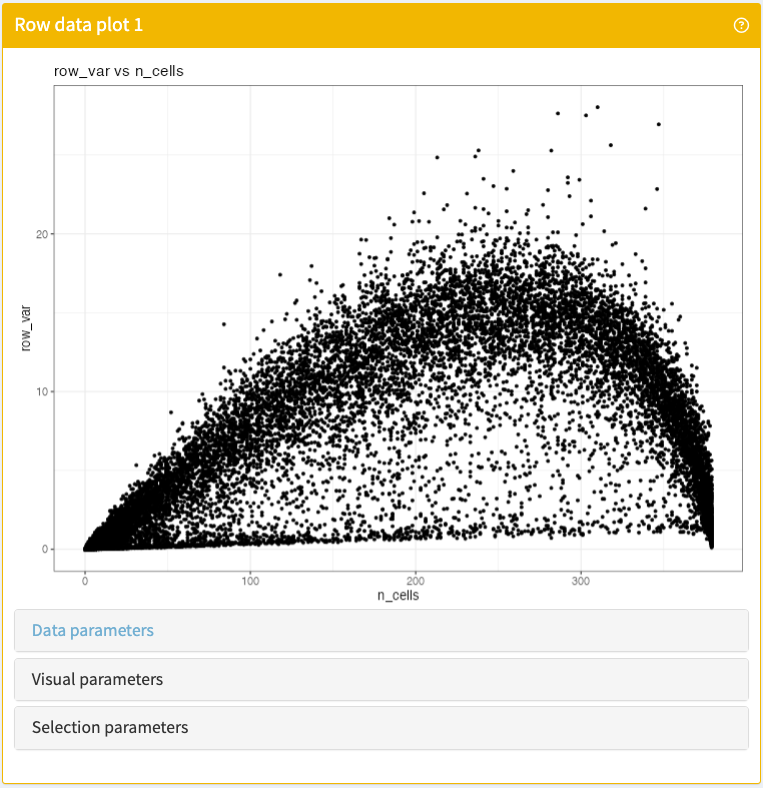
RowDataPlot panel class.
Reproduce This Output
library(iSEE)
library(scRNAseq)
library(scater)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
rowData(sce)$row_var <- rowVars(assay(sce, "logcounts"))
rowData(sce)$n_cells <- rowSums(assay(sce, "logcounts") > 0)
# launch the app itself ----
app <- iSEE(sce, initial = list(
RowDataPlot(
PanelWidth = 12L,
YAxis = "row_var",
XAxis = "Row data",
XAxisRowData = "n_cells"
)
))
if (interactive()) {
shiny::runApp(app, port=1234)
}RowDataTable
Browse and filter feature metadata stored in a SummarizedExperiment object.
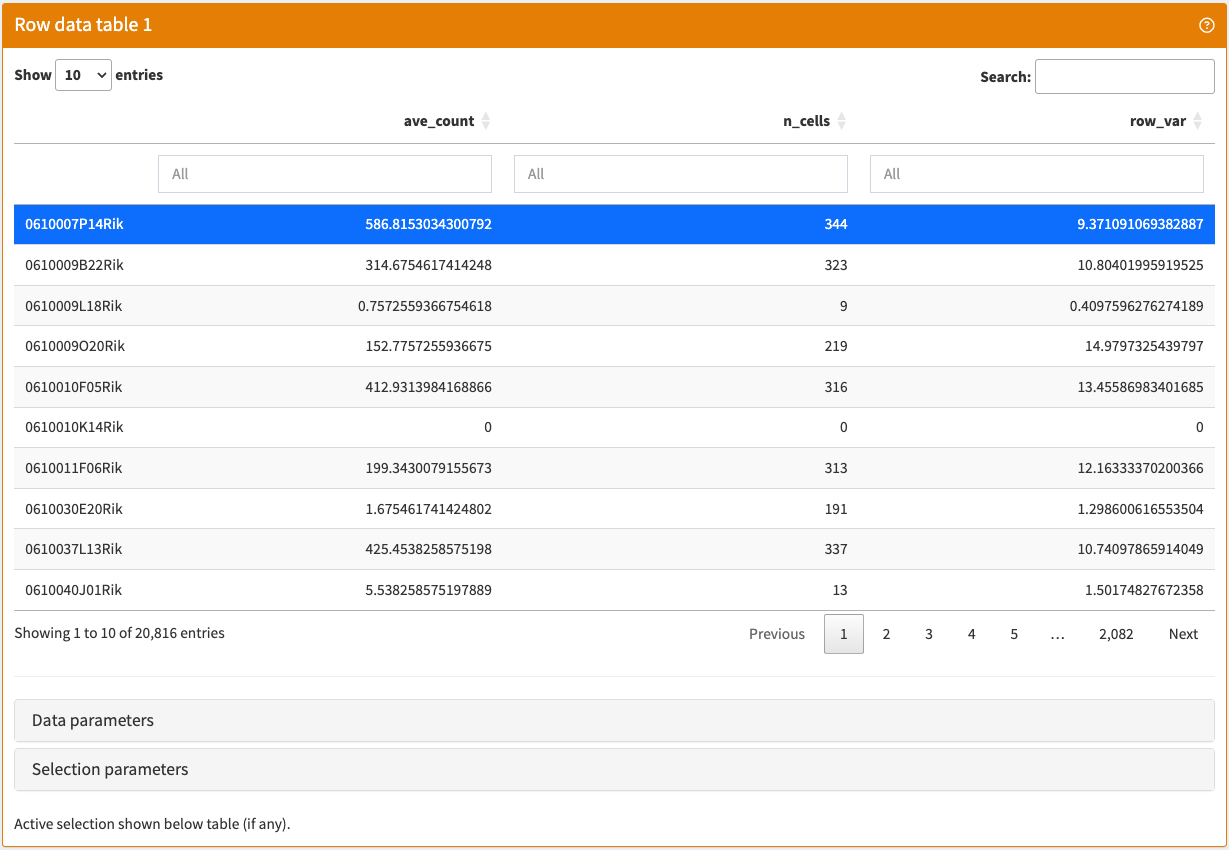
RowDataTable panel class.
Reproduce This Output
library(iSEE)
library(scRNAseq)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
rowData(sce)$ave_count <- rowMeans(assay(sce, "tophat_counts"))
rowData(sce)$n_cells <- rowSums(assay(sce, "tophat_counts") > 0)
# launch the app itself ----
app <- iSEE(sce, initial = list(
RowDataTable(
PanelWidth = 12L
)
))
if (interactive()) {
shiny::runApp(app, port=1234)
}SampleAssayPlot
Visualise up to two samples in any assay stored in a SummarizedExperiment object.
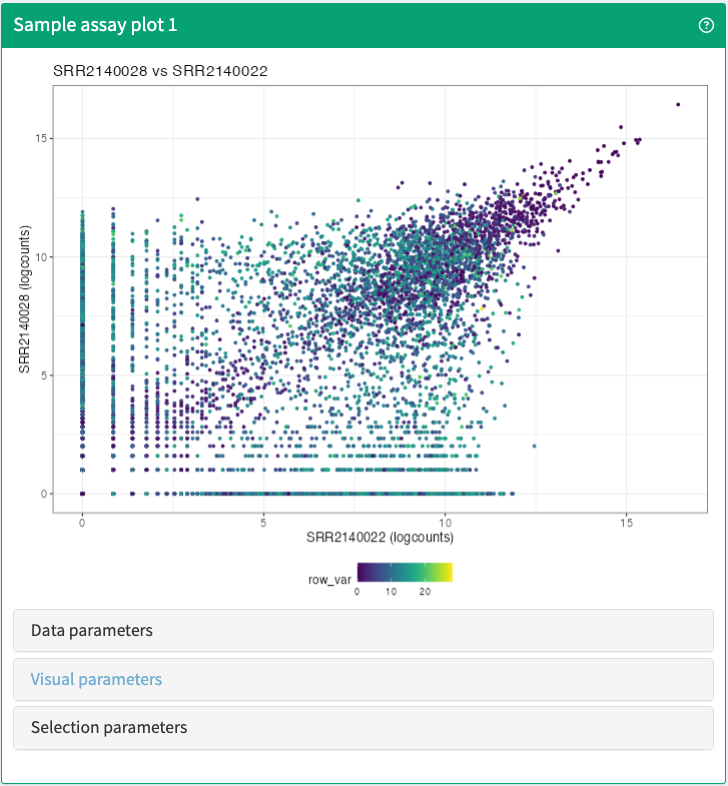
SampleAssayPlot panel class.
Reproduce This Output
library(iSEE)
library(scRNAseq)
library(scater)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
rowData(sce)$row_var <- rowVars(assay(sce, "logcounts"))
# launch the app itself ----
app <- iSEE(sce, initial = list(
SampleAssayPlot(
PanelWidth = 12L,
YAxisSampleName = "SRR2140028",
XAxis = "Sample name", XAxisSampleName = "SRR2140022",
ColorBy = "Row data", ColorByRowData = "row_var"
)
))
if (interactive()) {
shiny::runApp(app, port=1234)
}iSEEde
DETable
Browse and filter any table of differential expression results embedded in a SummarizedExperiment object.
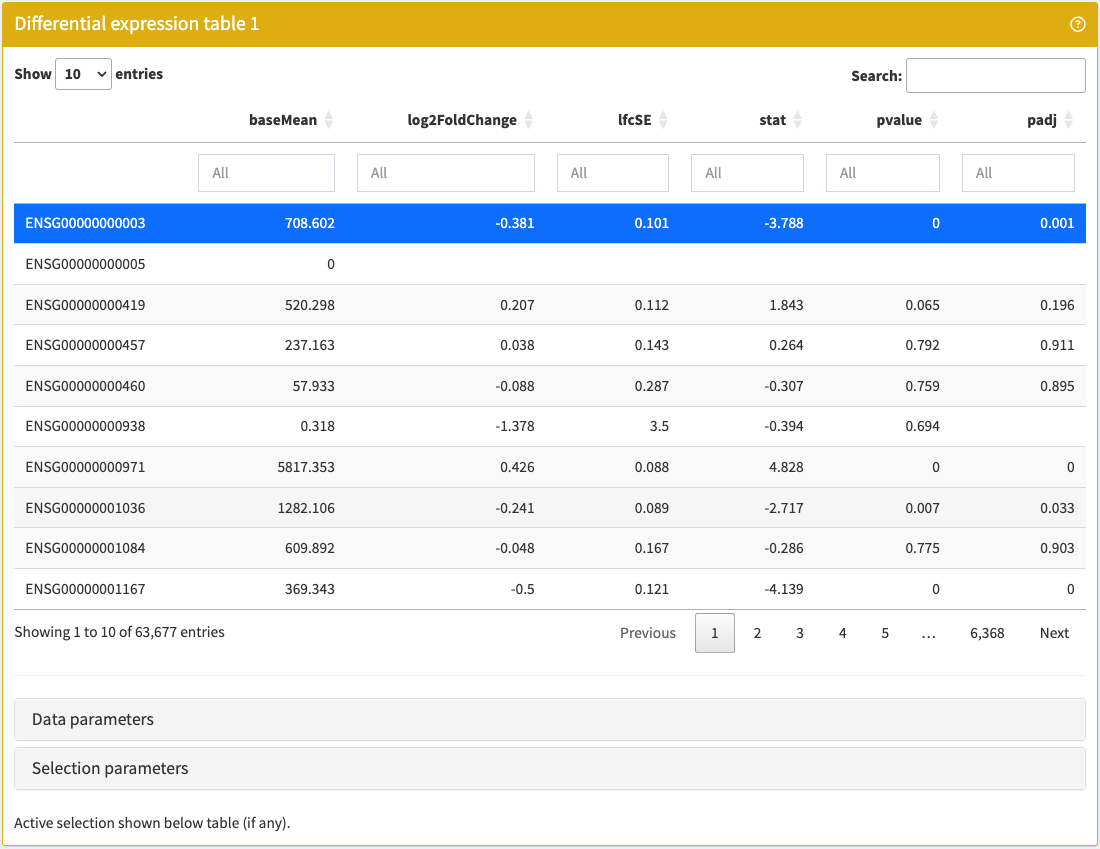
DETable panel class.
Reproduce This Output
library(iSEE)
library(iSEEde)
library(airway)
library(DESeq2)
# Example data ----
data("airway")
airway$dex <- relevel(airway$dex, "untrt")
dds <- DESeqDataSet(airway, ~ 0 + dex + cell)
dds <- DESeq(dds)
res_deseq2 <- results(dds, contrast = list("dextrt", "dexuntrt"))
# iSEE / iSEEde ---
airway <- embedContrastResults(res_deseq2, airway, name = "dex: trt vs untrt")
app <- iSEE(airway, initial = list(
DETable(
PanelWidth = 12L,
ContrastName="dex: trt vs untrt",
RoundDigits = TRUE
)
))
if (interactive()) {
shiny::runApp(app)
}LogFCLogFCPlot
Visualise the log-transformed fold-changes of any two differential expression contrasts embedded in a SummarizedExperiment object.
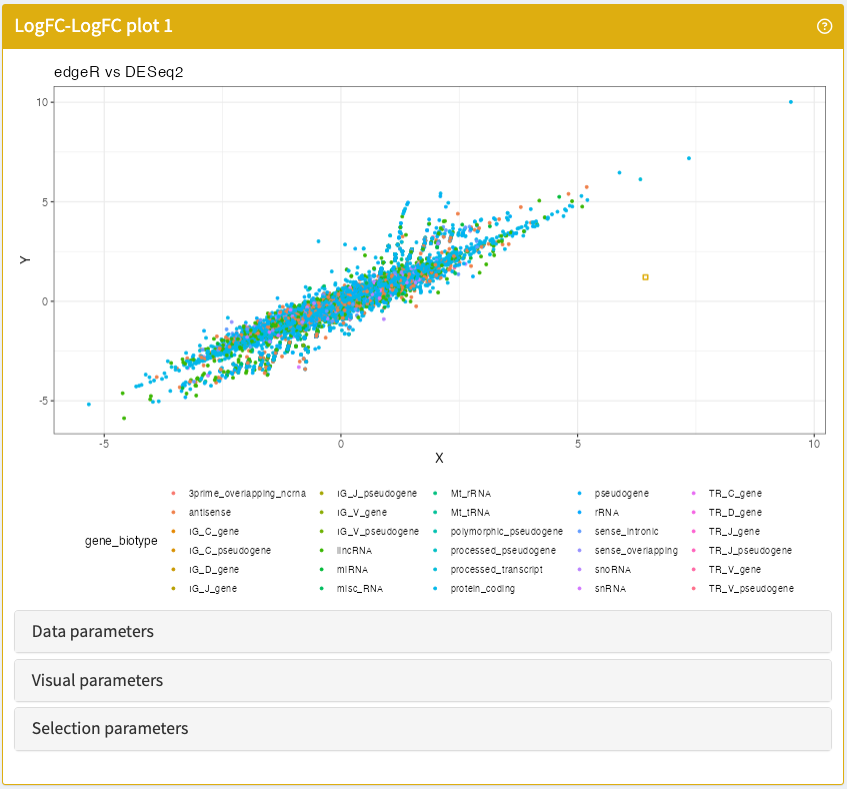
LogFCLogFCPlot panel class.
Reproduce This Output
library("iSEEde")
library("airway")
library("edgeR")
library("DESeq2")
library("iSEE")
# Example data ----
data("airway")
airway$dex <- relevel(airway$dex, "untrt")
# DESeq2 ----
dds <- DESeqDataSet(airway, ~ 0 + dex + cell)
dds <- DESeq(dds)
res_deseq2 <- results(dds, contrast = list("dextrt", "dexuntrt"))
airway <- embedContrastResults(res_deseq2, airway, name = "DESeq2")
# edgeR ----
design <- model.matrix(~ 0 + dex + cell, data = colData(airway))
fit <- glmFit(airway, design, dispersion = 0.1)
lrt <- glmLRT(fit, contrast = c(-1, 1, 0, 0, 0))
res_edger <- topTags(lrt, n = Inf)
airway <- embedContrastResults(res_edger, airway, name = "edgeR")
# iSEE / iSEEde ---
airway <- registerAppOptions(airway, factor.maxlevels = 30, color.maxlevels = 30)
app <- iSEE(airway, initial = list(
LogFCLogFCPlot(
ContrastNameX = "DESeq2", ContrastNameY = "edgeR",
ColorBy = "Row data",
ColorByRowData = "gene_biotype",
PanelWidth = 12L)
))
if (interactive()) {
shiny::runApp(app)
}MAPlot
Visualise the M and A values of any differential expression contrast embedded in a SummarizedExperiment object.
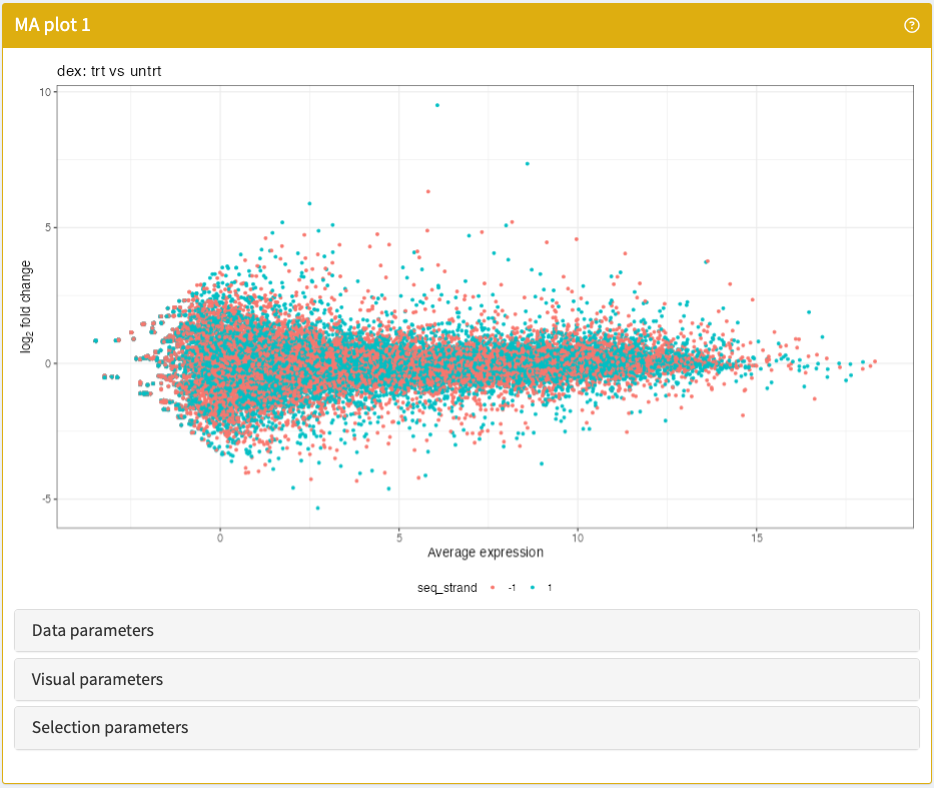
MAPlot panel class.
Reproduce This Output
library("iSEEde")
library("airway")
library("DESeq2")
library("iSEE")
# Example data ----
data("airway")
airway$dex <- relevel(airway$dex, "untrt")
rowData(airway)$seq_strand <- factor(rowData(airway)$seq_strand)
dds <- DESeqDataSet(airway, ~ 0 + dex + cell)
dds <- DESeq(dds)
res_deseq2 <- results(dds, contrast = list("dextrt", "dexuntrt"))
# iSEE / iSEEde ---
airway <- embedContrastResults(res_deseq2, airway, name = "dex: trt vs untrt")
app <- iSEE(airway, initial = list(
MAPlot(
PanelWidth = 12L,
ContrastName="dex: trt vs untrt",
ColorBy = "Row data",
ColorByRowData = "seq_strand"
)
))
if (interactive()) {
shiny::runApp(app)
}VolcanoPlot
Visualise the P values and log-transformed fold-changes of any differential expression contrast embedded in a SummarizedExperiment object.
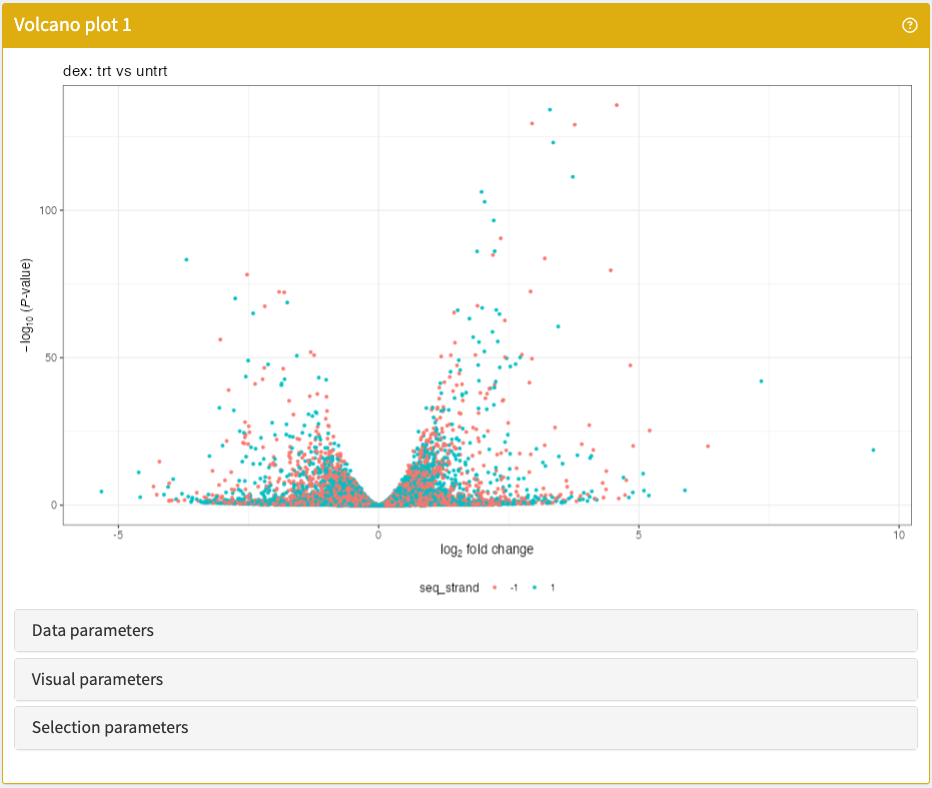
VolcanoPlot panel class.
Reproduce This Output
library(iSEE)
library(iSEEde)
library(airway)
library(DESeq2)
# Example data ----
data("airway")
airway$dex <- relevel(airway$dex, "untrt")
rowData(airway)$seq_strand <- factor(rowData(airway)$seq_strand)
dds <- DESeqDataSet(airway, ~ 0 + dex + cell)
dds <- DESeq(dds)
res_deseq2 <- results(dds, contrast = list("dextrt", "dexuntrt"))
# iSEE / iSEEde ---
airway <- embedContrastResults(res_deseq2, airway, name = "dex: trt vs untrt")
app <- iSEE(airway, initial = list(
VolcanoPlot(
PanelWidth = 12L,
ContrastName="dex: trt vs untrt",
ColorBy = "Row data",
ColorByRowData = "seq_strand"
)
))
if (interactive()) {
shiny::runApp(app)
}iSEEhex
ReducedDimensionHexPlot
Same as ReducedDimensionPlot but summarised using hexagonal bins.
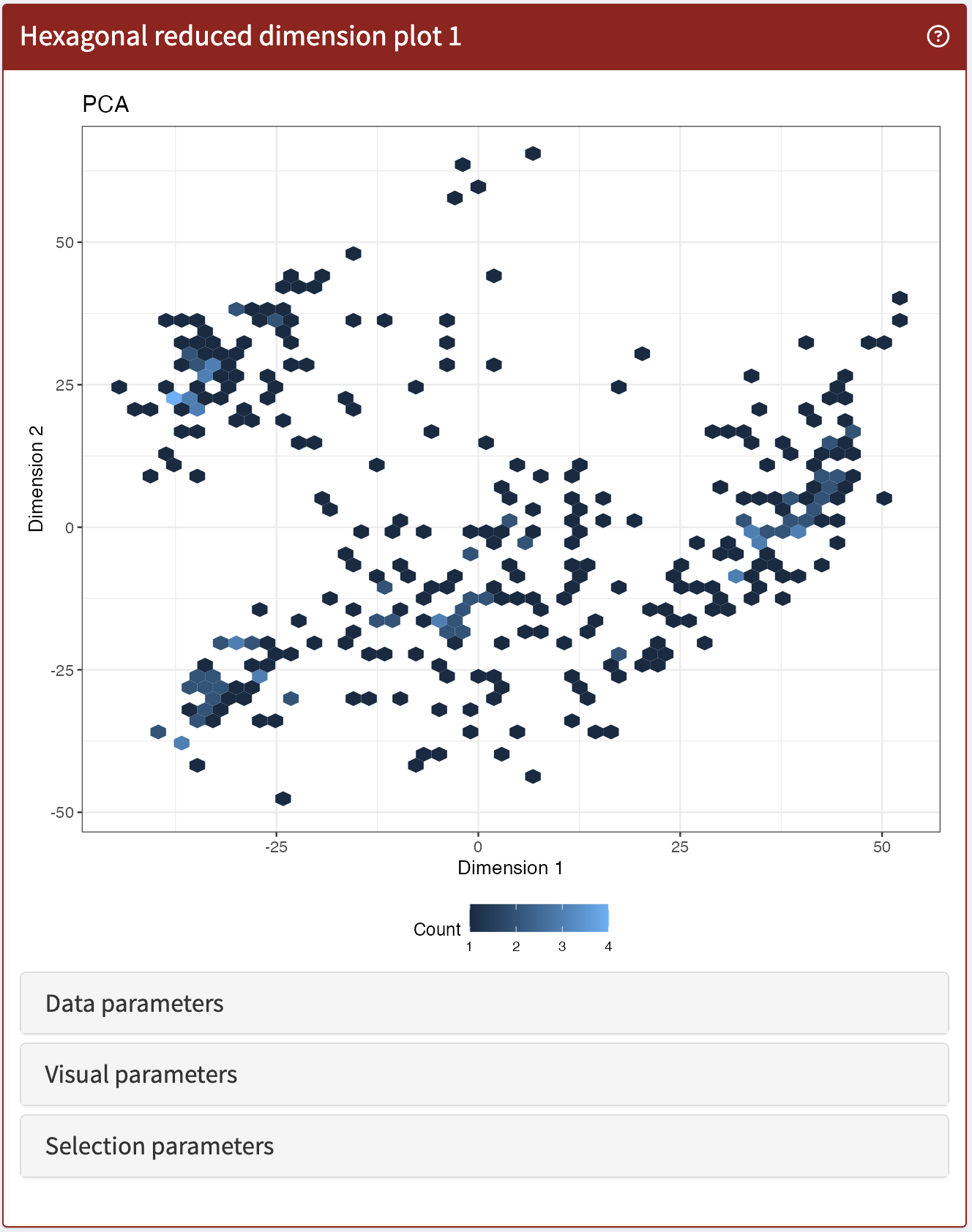
ReducedDimensionHexPlot panel class.
Reproduce This Output
library(iSEE)
library(iSEEhex)
library(scRNAseq)
library(scater)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
sce <- runPCA(sce, ncomponents=4)
# launch the app itself ----
if (interactive()) {
iSEE(sce, initial=list(
ReducedDimensionHexPlot(PanelWidth = 12L, BinResolution=50)
))
}iSEEpathways
FgseaEnrichmentPlot
GSEA enrichment plot produced by the fgsea package.
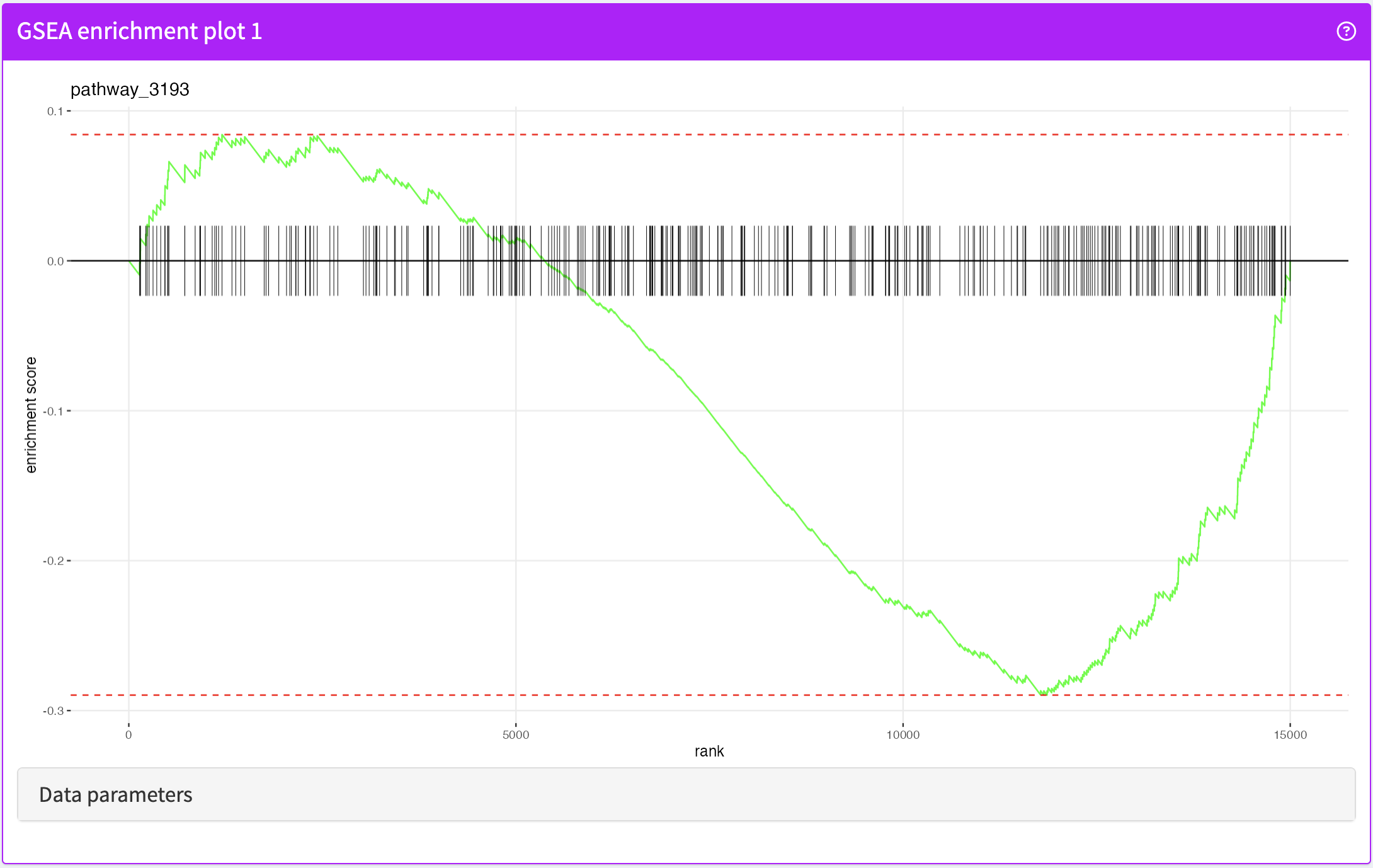
FgseaEnrichmentPlot panel class.
Reproduce This Output
library(iSEE)
library(fgsea)
library(iSEEpathways)
# Example data ----
set.seed(1)
simulated_data <- simulateExampleData()
pathways_list <- simulated_data[["pathwaysList"]]
features_stat <- simulated_data[["featuresStat"]]
se <- simulated_data[["summarizedexperiment"]]
# fgsea ----
set.seed(42)
fgseaRes <- fgsea(pathways = pathways_list,
stats = features_stat,
minSize = 15,
maxSize = 500)
fgseaRes <- fgseaRes[order(pval), ]
# iSEE / iSEEpathways ---
se <- embedPathwaysResults(fgseaRes, se, name = "fgsea", class = "fgsea", pathwayType = "simulated",
pathwaysList = pathways_list, featuresStats = features_stat)
app <- iSEE(se, initial = list(
FgseaEnrichmentPlot(ResultName="fgsea", PathwayId = "pathway_1350", PanelWidth = 12L)
))
if (interactive()) {
shiny::runApp(app)
}PathwaysTable
Browse and filter any table of gene set analysis results embedded in a SummarizedExperiment object.
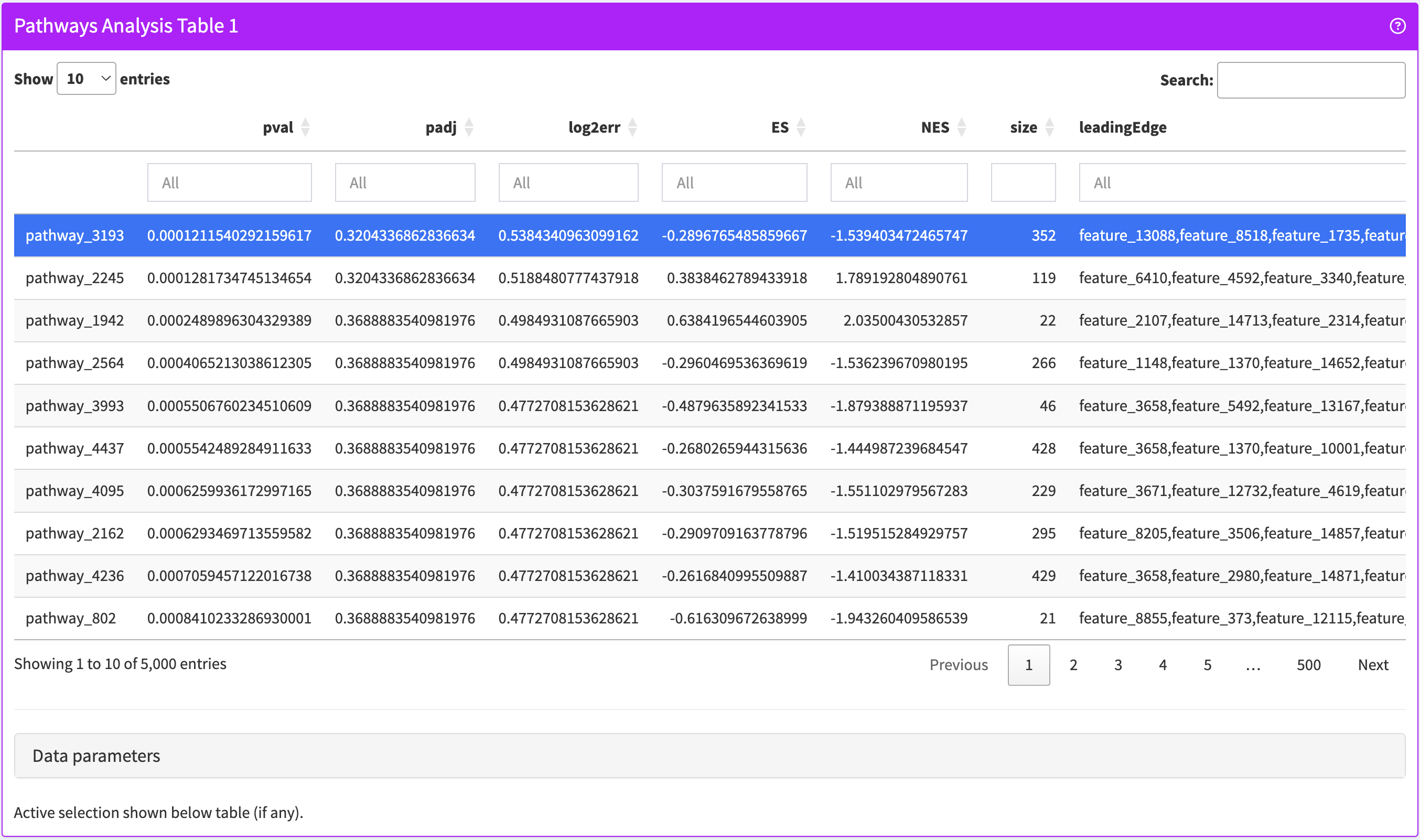
PathwaysTable panel class.
Reproduce This Output
library(iSEE)
library(fgsea)
library(iSEEpathways)
# Example data ----
set.seed(1)
simulated_data <- simulateExampleData()
pathways_list <- simulated_data[["pathwaysList"]]
features_stat <- simulated_data[["featuresStat"]]
se <- simulated_data[["summarizedexperiment"]]
# fgsea ----
set.seed(42)
fgseaRes <- fgsea(pathways = pathways_list,
stats = features_stat,
minSize = 15,
maxSize = 500)
fgseaRes <- fgseaRes[order(pval), ]
# iSEE ---
se <- embedPathwaysResults(fgseaRes, se, name = "fgsea", class = "fgsea", pathwayType = "simulated",
pathwaysList = pathways_list, featuresStats = features_stat)
app <- iSEE(se, initial = list(
PathwaysTable(ResultName="fgsea", PanelWidth = 12L)
))
if (interactive()) {
shiny::runApp(app)
}iSEEu
AggregatedDotPlot
Represents groups of samples by dots, where colour scales with means assay value and size scales with proportion of non-zero values for selected features.

AggregatedDotPlot panel class.
Reproduce This Output
library(iSEE)
library(scRNAseq)
library(scater)
library(iSEEu)
# Example data ----
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
# launch the app itself ----
if (interactive()) {
iSEE(
sce,
initial = list(
AggregatedDotPlot(
ColumnDataLabel="Primary.Type",
CustomRowsText = "Rorb\nSnap25\nFoxp2",
# PanelHeight = 500L,
PanelWidth = 12L
)
)
)
}DynamicMarkerTable
A table that dynamically identifies marker genes for a subset of samples transmitted from another panel. Comparisons are made between the active selection in the transmitting panel and either
- all non-selected points, if no saved selections are available, or
- each subset of points in each saved selection.

DynamicMarkerTable panel class, alongside a ReducedDimensionPlot panel from which it receives a selection of samples.
Reproduce This Output
library(iSEE)
library(iSEEu)
library(scRNAseq)
library(scater)
library(scran)
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
sce <- runPCA(sce, ncomponents=4)
if (interactive()) {
iSEE(sce, initial=list(
ReducedDimensionPlot(
PanelWidth=4L,
BrushData = list(
lasso = NULL, closed = TRUE,
mapping = list(x = "X", y = "Y"),
coord = structure(c(
-47.8, -41.9, -14.6, -13.6, -19.1, -27.3, -33.6, -44, -47.8,
-23.6, -44.1, -56.4, -46.9, -26.4, -17.4, -6.2, -5.4, -23.6),
dim = c(9L, 2L))
)
),
DynamicMarkerTable(
PanelWidth=8L,
ColumnSelectionSource="ReducedDimensionPlot1"
)
))
}DynamicReducedDimensionPlot
A dimensionality reduction plot that dynamically recomputes the coordinates for the samples, based on a subset of samples and features transmitted from other panels.

DynamicReducedDimensionPlot panel class, alongside a ReducedDimensionPlot panel from which it receives a selection of samples.
Reproduce This Output
library(iSEE)
library(iSEEu)
library(scRNAseq)
library(scater)
set.seed(1)
sce <- ReprocessedAllenData(assays="tophat_counts")
sce <- logNormCounts(sce, exprs_values="tophat_counts")
sce <- runPCA(sce, ncomponents=4)
if (interactive()) {
iSEE(sce, initial=list(
ReducedDimensionPlot(
PanelWidth = 6L,
BrushData = list(
lasso = NULL, closed = TRUE,
mapping = list(x = "X", y = "Y"),
coord = structure(c(
-47.8, -41.9, -14.6, -13.6, -19.1, -27.3, -33.6, -44, -47.8,
-23.6, -44.1, -56.4, -46.9, -26.4, -17.4, -6.2, -5.4, -23.6),
dim = c(9L, 2L))
)
),
DynamicReducedDimensionPlot(
PanelWidth = 6L,
Assay="logcounts",
ColumnSelectionSource="ReducedDimensionPlot1"
)
))
}FeatureSetTable
A table where each row is itself a set of features (i.e., rows) and can be used to transmit such a feature set to another panel.
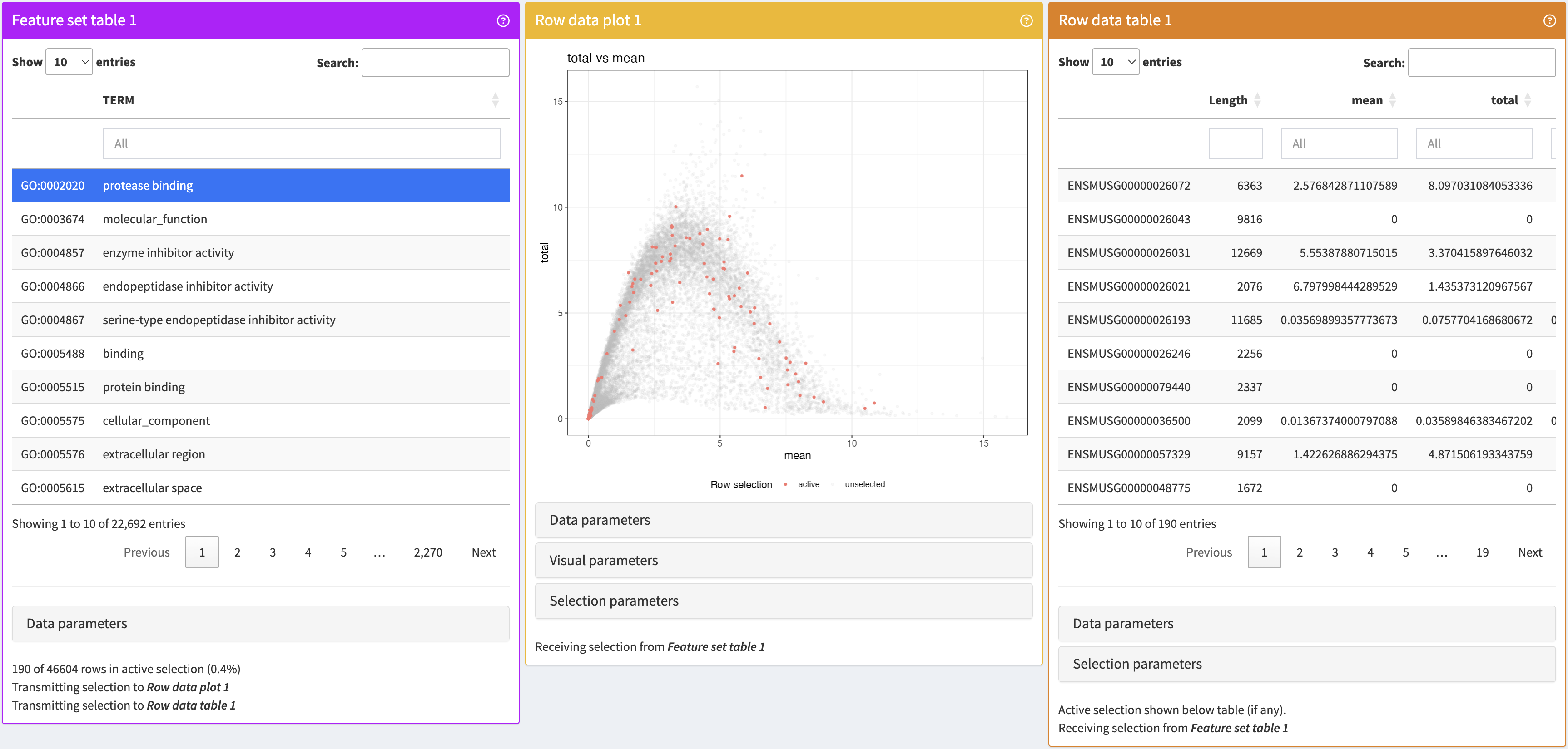
FeatureSetTable panel class, alongside RowDataPlot and RowDataTable panels to which it transmits a feature set.
Reproduce This Output
library(iSEE)
library(iSEEu)
library(scRNAseq)
library(scater)
library(scran)
library(org.Mm.eg.db)
sce <- LunSpikeInData(location=FALSE)
sce <- logNormCounts(sce)
rowData(sce) <- cbind(rowData(sce), modelGeneVarWithSpikes(sce, "ERCC"))
cmds <- createGeneSetCommands(collections="GO",
organism="org.Mm.eg.db", identifier="ENSEMBL")
sce <- registerFeatureSetCommands(sce, cmds)
# Setting up the application.
gst <- FeatureSetTable(
Selected = "GO:0002020"
)
rdp <- RowDataPlot(
YAxis="total",
XAxis="Row data", XAxisRowData="mean",
ColorBy="Row selection",
RowSelectionSource="FeatureSetTable1"
)
rdt <- RowDataTable(
RowSelectionSource="FeatureSetTable1"
)
if (interactive()) {
iSEE(sce, initial=list(gst, rdp, rdt))
}LogFCLogFCPlot
Precursor to the iSEEde::LogFCLogFCPlot class.
Warning
Deprecation imminent. Please use iSEEde::LogFCLogFCPlot() instead.

LogFCLogFCPlot panel class.
Reproduce This Output
# Making up some results:
se <- SummarizedExperiment(matrix(rnorm(10000), 1000, 10))
rownames(se) <- paste0("GENE_", seq_len(nrow(se)))
rowData(se)$PValue1 <- runif(nrow(se))
rowData(se)$LogFC1 <- rnorm(nrow(se))
rowData(se)$PValue2 <- runif(nrow(se))
rowData(se)$LogFC2 <- rnorm(nrow(se))
if (interactive()) {
iSEE(se, initial=list(
LogFCLogFCPlot(
PanelWidth = 12L,
XAxisRowData="LogFC1", YAxis="LogFC2",
XPValueField="PValue1", YPValueField="PValue2"
)
))
}MAPlot
Precursor to the iSEEde::MAPlot class.
Warning
Deprecation imminent. Please use iSEEde::MAPlot() instead.
MAPlot panel class.
Reproduce This Output
# Making up some results:
se <- SummarizedExperiment(matrix(rnorm(10000), 1000, 10))
rownames(se) <- paste0("GENE_", seq_len(nrow(se)))
rowData(se)$PValue <- runif(nrow(se))
rowData(se)$LogFC <- rnorm(nrow(se))
rowData(se)$AveExpr <- rnorm(nrow(se))
if (interactive()) {
iSEE(se, initial=list(
MAPlot(PanelWidth=12L)
))
}MarkdownBoard
A panel providing an aceEditor that can be used to take notes within the app. Notes should be typed using Markdown syntax, and the panel continuously renders them in HTML format for preview.

MarkdownBoard panel class.
Reproduce This Output
if (interactive()) {
iSEE(SummarizedExperiment(), initial=list(
MarkdownBoard(
PanelWidth = 12L,
Content = paste0(c(
"# Level 1 header",
"## Level 2 header",
"**Bold** and *italic*.",
"[Link](https://isee.github.io/)",
"* Bullet point\n"),
collapse = "\n\n"
)
)
))
}