Describing the ExperimentColorMap class
Kevin Rue-Albrecht
MRC WIMM Centre for Computational Biology, University of Oxford, Oxford, OX3 9DS, UKkevinrue67@gmail.com
Federico Marini
Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI), MainzCenter for Thrombosis and Hemostasis (CTH), Mainzmarinif@uni-mainz.de
Charlotte Soneson
Friedrich Miescher Institute for Biomedical Research, Basel, SwitzerlandSIB Swiss Institute of Bioinformaticscharlottesoneson@gmail.com
Aaron Lun
infinite.monkeys.with.keyboards@gmail.com13 July 2026
Source:vignettes/ecm.Rmd
ecm.RmdCompiled date: 2026-07-13
Last edited: 2018-03-08
License: MIT + file LICENSE
Background
iSEE
coordinates the coloration in every plot via the
ExperimentColorMap class (Rue-Albrecht et al. 2018). Colors for samples
or features are defined from column or row metadata or assay values
using “colormaps”. Each colormap is a function that takes a single
integer argument and returns that number of distinct colors. The
ExperimentColorMap is a container that stores these
functions for use within the iSEE() function. Users can
define their own colormaps to customize coloration for specific assays
or covariates.
Defining colormaps
Colormaps for continuous variables
For continuous variables, the function will be asked to generate a
number of colors (21, by default). Interpolation will then be performed
internally to generate a color gradient. Users can use existing color
scales like viridis::viridis or
heat.colors:
# Coloring for log-counts:
logcounts_color_fun <- viridis::viridisIt is also possible to use a function that completely ignores any arguments, and simply returns a fixed number of interpolation points:
# Coloring for FPKMs:
fpkm_color_fun <- function(n){
c("black","brown","red","orange","yellow")
}Colormaps for categorical variables
For categorical variables, the function should accept the number of levels and return a color per level. Colors are automatically assigned to factor levels in the specified order of the levels.
# Coloring for the 'driver' metadata variable.
driver_color_fun <- function(n){
RColorBrewer::brewer.pal(n, "Set2")
}Alternatively, the function can ignore its arguments and simply
return a named vector of colors if users want to specify the color for
each level explicitly It is the user’s responsibility to ensure that all
levels are accounted for1. For instance, the following colormap
function will only be compatible with factors of two levels, namely
"Y" and "N":
The colormap hierarchy
Specific and shared colormaps
Colormaps can be defined by users at three different levels:
- Each individual assay, column data field, and row data field can be
assigned its own distinct colormap. Those colormaps are stored as named
lists of functions in the
assays,colData, androwDataslots, respectively, of theExperimentColorMap. This can be useful to easily remember which assay is currently shown; to apply different color scale limits to assays that vary on different ranges of values; or display boolean information in an intuitive way, among many other scenarios. -
Shared colormaps can be defined for all assays, all column
data, and all row data. These colormaps are stored in the
all_discreteandall_continuousslots of theExperimentColorMap, as lists of functions namedassays,colData, androwData. -
Global colormaps can be defined for all categorical or
continuous data. Those two colormaps are stored in the
global_discreteandglobal_continuousslots of theExperimentColorMap.
Searching for colors
When queried for a specific colormap of any type (assay, column data, or row data), the following process takes place:
- A specific individual colormap is looked up in the
appropriate slot of the
ExperimentColorMap. - If it is not found, the shared colormap of the appropriate slot is looked up, according to whether the data are categorical or continuous.
- If it is not found, the global colormap is looked up, according to whether the data are categorical or continuous.
- If none of the above colormaps were defined, the
ExperimentColorMapwill revert to the default colormaps.
By default, viridis is used as the default continuous
colormap, and hcl is used as the default categorical
colormap.
Creating the ExperimentColorMap
We store the set of colormap functions in an instance of the
ExperimentColorMap class. Named functions passed as
assays, colData, or rowData
arguments will be used for coloring data in those slots,
respectively.
library(iSEE)
ecm <- ExperimentColorMap(
assays = list(
counts = heat.colors,
logcounts = logcounts_color_fun,
cufflinks_fpkm = fpkm_color_fun
),
colData = list(
passes_qc_checks_s = qc_color_fun,
driver_1_s = driver_color_fun
),
all_continuous = list(
assays = viridis::plasma
)
)
ecm
#> Class: ExperimentColorMap
#> assays(3): counts logcounts cufflinks_fpkm
#> colData(2): passes_qc_checks_s driver_1_s
#> rowData(0):
#> all_discrete(0):
#> all_continuous(1): assaysUsers can change the defaults for all assays or column data by modifying the shared colormaps. Similarly, users can modify the defaults for all continuous or categorical data by modifying the global colormaps. This is demonstrated below for the continuous variables:
ExperimentColorMap(
all_continuous=list( # shared
assays=viridis::plasma,
colData=viridis::inferno
),
global_continuous=viridis::magma # global
)
#> Class: ExperimentColorMap
#> assays(0):
#> colData(0):
#> rowData(0):
#> all_discrete(0):
#> all_continuous(2): assays colData
#> global_continuous(1)Benefits
The ExperimentColorMap class offers the following major
features:
- A single place to define flexible and lightweight sets of colormaps, that may be saved and reused across sessions and projects outside the app, to apply consistent coloring schemes across entire projects
- A simple interface through accessors
colDataColorMap(colormap, "coldata_name")and settersassayColorMap(colormap, "assay_name") <- colormap_function - An elegant fallback mechanism to consistently return a colormap, even for undefined covariates, including a default categorical and continuous colormap, respectively.
- Three levels of colormaps override: individual, shared within slot
(i.e.,
assays,colData,rowData), or shared globally between all categorical or continuous data scales.
Detailed examples on the use of ExperimentColorMap
objects are available in the documentation
?ExperimentColorMap, as well as below.
Demonstration
Here, we use the allen single-cell RNA-seq data set to
demonstrate the use of the ExperimentColorMap class. Using
the sce object that we created previously,
we create an iSEE app with the
SingleCellExperiment object and the colormap generated
above.
app <- iSEE(sce, colormap = ecm)We run this using runApp to open the app on our
browser.
shiny::runApp(app)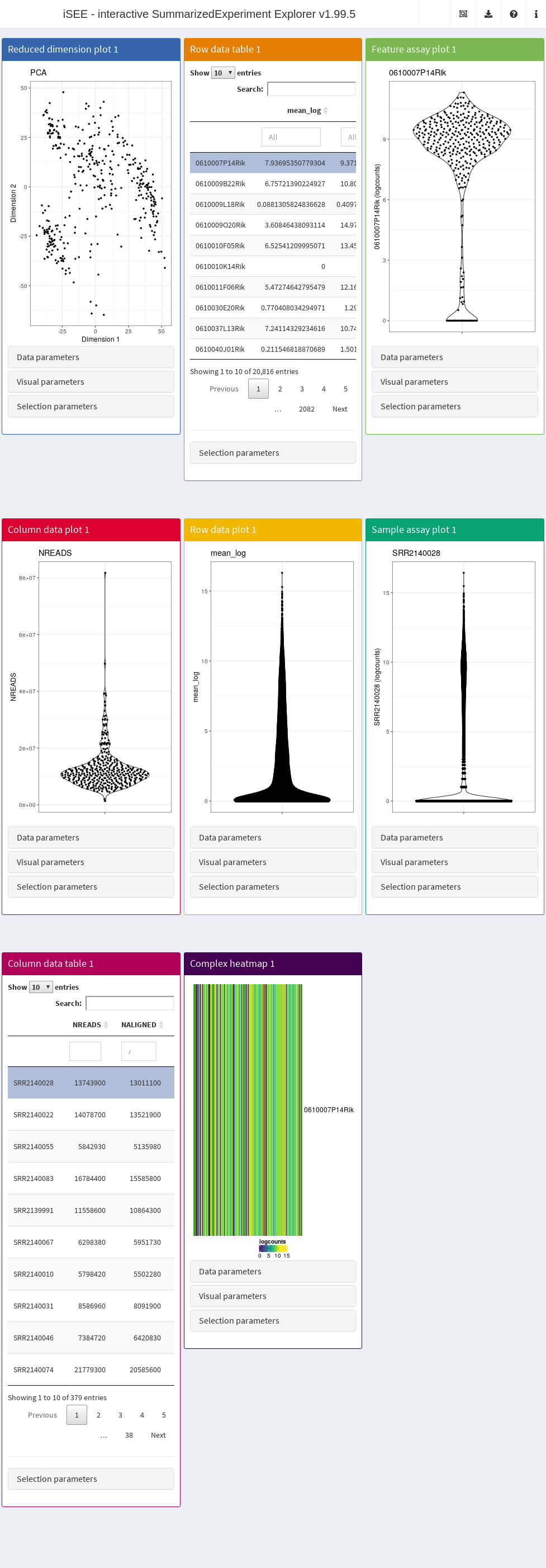
Now, choose to color cells by Column data and select
passes_qc_checks_s. We will see that all cells that passed
QC (Y) are colored “forestgreen”, while the ones that
didn’t pass are colored firebrick.
If we color any plot by gene expression, we see that use of counts
follows the heat.colors coloring scheme; use of log-counts
follows the viridis coloring scheme; and use of FPKMs
follows the black-to-yellow scheme we defined in
fpkm_color_fun.
Session Info
sessionInfo()
#> R version 4.6.1 (2026-06-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] iSEE_2.25.1 SingleCellExperiment_1.34.0
#> [3] SummarizedExperiment_1.42.0 Biobase_2.72.0
#> [5] GenomicRanges_1.64.0 Seqinfo_1.2.0
#> [7] IRanges_2.46.0 S4Vectors_0.50.1
#> [9] BiocGenerics_0.58.1 generics_0.1.4
#> [11] MatrixGenerics_1.24.0 matrixStats_1.5.0
#> [13] BiocStyle_2.40.0
#>
#> loaded via a namespace (and not attached):
#> [1] gridExtra_2.3.1 rlang_1.3.0 magrittr_2.0.5
#> [4] shinydashboard_0.7.3 clue_0.3-68 GetoptLong_1.1.1
#> [7] otel_0.2.0 compiler_4.6.1 mgcv_1.9-4
#> [10] png_0.1-9 systemfonts_1.3.2 vctrs_0.7.3
#> [13] pkgconfig_2.0.3 shape_1.4.6.1 crayon_1.5.3
#> [16] fastmap_1.2.0 XVector_0.52.0 fontawesome_0.5.3
#> [19] promises_1.5.0 rmarkdown_2.31 shinyAce_0.4.4
#> [22] ragg_1.5.2 xfun_0.60 cachem_1.1.0
#> [25] jsonlite_2.0.0 listviewer_4.0.0 later_1.4.8
#> [28] DelayedArray_0.38.2 parallel_4.6.1 cluster_2.1.8.2
#> [31] R6_2.6.1 bslib_0.11.0 RColorBrewer_1.1-3
#> [34] jquerylib_0.1.4 Rcpp_1.1.2 bookdown_0.47
#> [37] iterators_1.0.14 knitr_1.51 httpuv_1.6.17
#> [40] Matrix_1.7-5 splines_4.6.1 igraph_2.3.3
#> [43] tidyselect_1.2.1 abind_1.4-8 yaml_2.3.12
#> [46] viridis_0.6.5 doParallel_1.0.17 codetools_0.2-20
#> [49] miniUI_0.1.2 lattice_0.22-9 tibble_3.3.1
#> [52] shiny_1.14.0 S7_0.2.2 evaluate_1.0.5
#> [55] desc_1.4.3 circlize_0.4.18 pillar_1.11.1
#> [58] BiocManager_1.30.27 DT_0.34.0 foreach_1.5.2
#> [61] shinyjs_2.1.1 ggplot2_4.0.3 scales_1.4.0
#> [64] xtable_1.8-8 glue_1.8.1 tools_4.6.1
#> [67] colourpicker_1.3.0 fs_2.1.0 grid_4.6.1
#> [70] colorspace_2.1-3 nlme_3.1-169 vipor_0.4.7
#> [73] cli_3.6.6 textshaping_1.0.5 S4Arrays_1.12.0
#> [76] viridisLite_0.4.3 ComplexHeatmap_2.28.0 dplyr_1.2.1
#> [79] gtable_0.3.6 rintrojs_0.3.4 sass_0.4.10
#> [82] digest_0.6.39 SparseArray_1.12.2 ggrepel_0.9.8
#> [85] rjson_0.2.23 htmlwidgets_1.6.4 farver_2.1.2
#> [88] memoise_2.0.1 htmltools_0.5.9 pkgdown_2.2.1
#> [91] lifecycle_1.0.5 shinyWidgets_0.9.1 GlobalOptions_0.1.4
#> [94] mime_0.13
# devtools::session_info()