An introduction to the iSEE interface
Kevin Rue-Albrecht
MRC WIMM Centre for Computational Biology, University of Oxford, Oxford, OX3 9DS, UKkevinrue67@gmail.com
Federico Marini
Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI), MainzCenter for Thrombosis and Hemostasis (CTH), Mainzmarinif@uni-mainz.de
Charlotte Soneson
Friedrich Miescher Institute for Biomedical Research, Basel, SwitzerlandSIB Swiss Institute of Bioinformaticscharlottesoneson@gmail.com
Aaron Lun
infinite.monkeys.with.keyboards@gmail.com13 July 2026
Source:vignettes/basic.Rmd
basic.RmdCompiled date: 2026-07-13
Last edited: 2020-04-20
License: MIT + file LICENSE
Introduction
iSEE is a Bioconductor package that provides an
interactive Shiny-based graphical user interface for exploring data
stored in SummarizedExperiment objects (Rue-Albrecht et al. 2018). Instructions to
install the package are available here. Once
installed, the package can be loaded and attached to your current
workspace as follows:
If you have a SummarizedExperiment object1 named se,
you can launch an iSEE app by
running:
iSEE(se)In this vignette, we demonstrate this process using the
allen single-cell RNA-seq data set from the scRNAseq
package. However, if you want to start playing with the app immediately,
you can simply run:
example(iSEE, ask=FALSE)Setting up the data
The allen data set contains expression values for 379
cells from the mouse visual cortex (Tasic et al.
2016), and can be loaded directly by calling
ReprocessedAllenData() and specifying the value for the
assays parameter. To begin with, we assign the output of
this call to an sce object and inspect it.
library(scRNAseq)
sce <- ReprocessedAllenData(assays = "tophat_counts") # specifying the assays to speed up the example
sce
#> class: SingleCellExperiment
#> dim: 20816 379
#> metadata(2): SuppInfo which_qc
#> assays(1): tophat_counts
#> rownames(20816): 0610007P14Rik 0610009B22Rik ... Zzef1 Zzz3
#> rowData names(0):
#> colnames(379): SRR2140028 SRR2140022 ... SRR2139341 SRR2139336
#> colData names(22): NREADS NALIGNED ... Animal.ID passes_qc_checks_s
#> reducedDimNames(0):
#> mainExpName: endogenous
#> altExpNames(1): ERCCAs provided, the sce object contains raw data and a
number of quality control and experimental cell annotations, all
available in colData(sce).
colnames(colData(sce))
#> [1] "NREADS" "NALIGNED"
#> [3] "RALIGN" "TOTAL_DUP"
#> [5] "PRIMER" "PCT_RIBOSOMAL_BASES"
#> [7] "PCT_CODING_BASES" "PCT_UTR_BASES"
#> [9] "PCT_INTRONIC_BASES" "PCT_INTERGENIC_BASES"
#> [11] "PCT_MRNA_BASES" "MEDIAN_CV_COVERAGE"
#> [13] "MEDIAN_5PRIME_BIAS" "MEDIAN_3PRIME_BIAS"
#> [15] "MEDIAN_5PRIME_TO_3PRIME_BIAS" "driver_1_s"
#> [17] "dissection_s" "Core.Type"
#> [19] "Primary.Type" "Secondary.Type"
#> [21] "Animal.ID" "passes_qc_checks_s"Then, we normalize the expression values with scrapper.
library(scrapper)
sce <- normalizeRnaCounts.se(sce, assay.type = "tophat_counts")Next, we apply PCA and t-SNE to generate two low-dimensional
representations of the cells. The dimensionality reduction results are
stored in reducedDim(sce).
set.seed(1000)
library(scater)
sce <- runPCA(sce)
sce <- runTSNE(sce)
reducedDimNames(sce)
#> [1] "PCA" "TSNE"At this point, the sce object does not contain any
annotations for the rows (i.e., features) in the data set. Thus, to
prepare a fully-featured example application, we also add some gene
metadata to the rowData related to the mean-variance
relationship in the data.
rowData(sce)$mean_log <- rowMeans(logcounts(sce))
rowData(sce)$var_log <- apply(logcounts(sce), 1, var)It is important to note that iSEE relies primarily on
precomputed values stored in the various slots of objects derived from
the SummarizedExperiment class2. This allows users to
visualize any metrics of interest, but also requires these to be
calculated and added to the object before the initialization of the app.
That said, it is straightforward to iteratively explore a precomputed
object, take notes of new metrics to compute, close the app, store new
results in the SummarizedExperiment object, and launch a
new app using the updated object.
Launching the interface
To begin the exploration, we create an iSEE app with the
SingleCellExperiment object generated above. In its
simplest form, the iSEE function only requires the input
object. However, iSEE
applications can be extensively reconfigured
using a number of optional arguments to the iSEE
function.
app <- iSEE(sce)The runApp function launches the app in our browser.
shiny::runApp(app)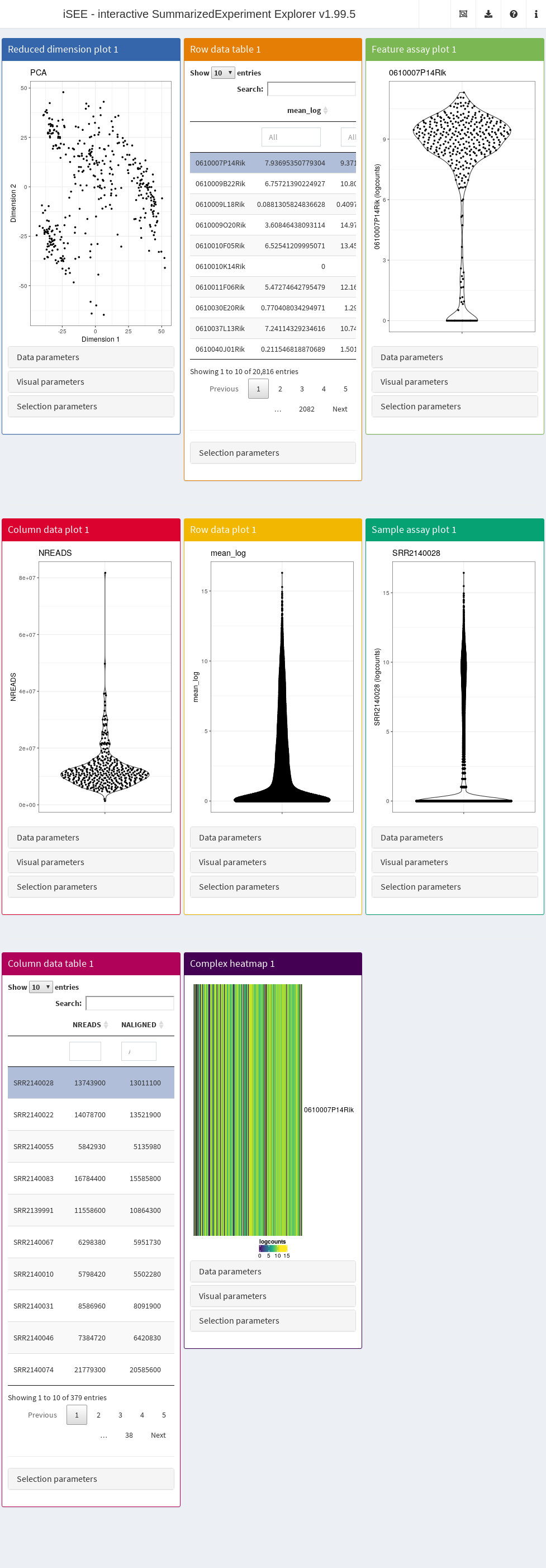
By default, the app starts with a dashboard that contains one panel or table of each type. By opening the collapsible panels named “Data parameters”, “Visual parameters”, and “Selection parameters” under each plot, we can control the content and appearance of each panel.
Now, look in the upper right corner for a question mark icon (), and click on the hand button () for an introductory tour. This will perform an interactive tour of the app, based on the rintrojs package (Ganz 2016). During this tour, you will be taken through the different components of the iSEE user interface and learn the basic usage mechanisms by doing small actions guided by the tutorial: the highlighted elements will be responding to your actions, while the rest of the UI will be shaded. You can move forward and backward along the tour by clicking on the “Next”/“Back” buttons, or also using the arrow keys. You can even jump to a particular step by clicking on its circle. To exit the tour, either click on “Skip”, or simply click outside of the highlighted UI element.
Once you are done generating plots, click on the export icon () in the upper right corner, and click on the magic wand button () to display R code that you can export and directly re-use in your R session. This will open a modal popup where the R code used to generate the plots is displayed in a shinyAce-based text editor. Select parts or all of it to copy-and-paste it into your analysis script/Rmarkdown file. However, note that the order in which the code blocks are reported is important if you have linked panels to one another, as the panels sending point selections must be executed before those that receive the corresponding selection.
Description of the user interface
Header
The layout of the iSEE user interface uses the shinydashboard package. The dashboard header contains four dropdown menus.
The first icon in the dashboard header is the “Organization” menu, which is identified by an icon displaying multiple windows (). This menu contains two items:
- The “Organize panels” button. Click on this button to open a modal
window that contains:
- A selectize input to add, remove, and reorder panels in the main interface.
- Two inputs to control the width and height, respectively, of each
panel selected above. Note that panel width is defined in integer grid
units between 1 and 12 (see
?shiny::column). In contrast, panel height is defined in pixels (see?shiny::plotOutput).
- The “Examine panel chart” functionality, identified by a chain icon (). Click on this button to obtain a graph representation of the existing links and point selections among your visible plot and table panels. Every panel is represented by a node coded with the same color as in the app. This can be very useful in sessions that include a large number of panels, to visualize the relationship structure between the various panels that send and receive selections of data points.
The second icon in the dashboard header is the “Export” dropdown menu, which is identified by a download icon () and contains:
- The “Download panel output” functionality, identified by the download icon (). Here, you can download a zip folder with the currently displayed panel content (high-resolution figures and table contents as csv files).
- The “Extract the R code”, functionality
(). At any point during your live session,
you might want to record the code that reproduces exactly the state of
each plot. Clicking on this button opens a modal popup window, with a
shinyAce-based
text editor, where the code is formatted and displayed with syntax
highlighting. You can copy the code to the clipboard by selecting the
text (please do include the initial lines and the
sessionInfo()commands for best tracking of your environment), and store it in your analysis report/script. This code can then be further edited to finalize the plots (e.g., for publication). - The “Display panel settings” () button, which lets you export the code defining the state of the current panels in the interface. This is useful if you want to preconfigure an iSEE instance to start in the current state, rather than with the default set of panels.
The “Documentation” dropdown menu is accessible through the question mark icon (), which contains:
- The button to start an interactive tour () of iSEE, which allows users to learn the basic usage mechanisms by doing. During a tour, the highlighted elements respond to the user’s actions, while the rest of the UI is shaded.
- The button to “Open the vignette” (), which displays the iSEE vignette, either available on your system or accessed at the webpage of the package on the Bioconductor project site. In the latter case, the vignette will refer to the current release or development version, according to the version of the package installed on your system).
The “Additional Information” dropdown menu is accessible through the information icon (), and contains:
- The “About this session” button
(), which reports the output of the
sessionInfo()function in a modal popup window. This is particularly useful for reproducing or reporting the environment, especially when reporting errors or unexpected behaviors. - The “About iSEE” button () shows the information on the development team, licensing and citation information for the iSEE package. You can follow the development of the package by checking the GitHub repository (https://github.com/iSEE/iSEE), where new functionality will be added. Well-considered suggestions in the form of issues or pull requests are welcome.
Body
Overview of panel types
The main element in the body of iSEE is the combination of panels, generated (and optionally linked to one another) according to your actions. There are currently eight standard panel types that can be generated with iSEE:
- Reduced dimension plot
- Column data table
- Column data plot
- Feature assay plot
- Row data table
- Row data plot
- Sample assay plot
- Complex heatmap
In addition, custom panel types can be defined as described in a separate dedicated vignette. The panels and models in the iSEEu package provide additional flexibility.
For each standard plot panel, three different sets of parameters will be available in collapsible boxes:
- “Data parameters”, to control parameters specific to each type of plot.
- “Visual parameters”, to specify parameters that will determine the aspect of the plot, in terms of coloring, point features, and more (e.g., legend placement, font size).
- “Selection parameters” to control the incoming point selection and link relationships to other plots.
Reduced dimension plots
If a SingleCellExperiment object is supplied to the
iSEE function, reduced dimension results are extracted from
the reducedDim slot. Examples include low-dimensional
embeddings from principal components analysis (PCA) or
t-distributed stochastic neighbour embedding (t-SNE)
(Van der Maaten and Hinton 2008). These
results are used to construct a two-dimensional Reduced dimension
plot where each point is a sample, to facilitate efficient
exploration of high-dimensional datasets. The “Data parameters” control
the reducedDim slot to be displayed, as well as the two
dimensions to plot against each other.
Note that this builtin panel does not compute reduced dimension
embeddings; they must be precomputed and available in the object
provided to the iSEE function. Nevertheless, custom panels
- such as the iSEEu
DynamicReducedDimensionPlot can be developed and used to
enable such features.
Column data plots
A Column data plot visualizes sample metadata stored in the
SummarizedExperiment column metadata. Different fields can
be used for the x- and y-axes by selecting appropriate values in the
“Data parameters” box. This plot can assume various forms, depending on
the nature of the data on the x- and y-axes:
- If the y-axis is continuous and the x-axis is categorical, violin plots are generated (grouped by the x-axis factor).
- If the y-axis is categorical and the x-axis is continuous, horizontal violin plots are generated (grouped by the y-axis factor).
- If both axes are continuous, a scatter plot is generated. This
enables the use of contours that are overlaid on top of the plot, check
the
"Other"box to see the available options. - If both axes are categorical, a plot of squares (Hinton plot) is generated where the area of each square is proportional to the number of samples within each combination of factor levels.
Note that an x-axis setting of “None” is considered to be categorical with a single level.
Feature assay plots
A Feature assay plot visualizes the assayed values (e.g.,
gene expression) for a particular feature (e.g., gene) across the
samples on the y-axis. This usually results in a (grouped) violin plot,
if the x-axis is set to "None" or a categorical variable;
or a scatter plot, if the x-axis is another continuous variable3.
Gene selection for the y-axis can be achieved by using a linked
row data table in another panel. Clicking on a row in the table
automatically changes the assayed values plotted on the y-axis.
Alternatively, the row name can be directly entered as text that
corresponds to an entry of rownames(se)4.
The x-axis covariate can also be selected from the plotting
parameters. This can be "None", sample metadata, or the
assayed values of another feature (also identified using a linked table
or via text). The measurement units are selected as one of the
assays(se), which is applied to both the X and Y axes.
Obviously, any other assayed value for any feature can be visualized
in this manner, not limited to the expression of genes. The only
requirement for this type of panel is that the observations can be
stored as a matrix in the SummarizedExperiment object.
Row data plots
A Row data plot allows the visualization of information
stored in the rowData slot of a
SummarizedExperiment object. Its behavior mirrors the
implementation for the Column data plot, and correspondingly
this plot can assume various forms depending on whether the data are
categorical or continuous.
Sample assay plots
A Sample assay plot visualizes the assayed values (e.g., gene expression) for a particular sample (e.g., cell) across the features on the y-axis.
This usually results in a (grouped) violin plot, if the x-axis is set
to "None" or a categorical variable (e.g., gene biotype);
or a scatter plot, if the x-axis is another continuous variable.
Notably, the x-axis covariate can also be set to:
- A discrete row data covariates (e.g., gene biotype), to stratify the distribution of assayed values
- A continuous row data covariate (e.g., count of cells expressing each gene)
- Another sample, to visualize and compare the assayed values in any two samples.
Row data tables
A Row data table contains the values of the
rowData slot for the
SingleCellExperiment/SummarizedExperiment
object. If none are available, a column named Present is
added and set to TRUE for all features, to avoid issues
with DT::datatable and an empty DataFrame.
Typically, these tables are used to link to other plots to determine the
features to use for plotting or coloring. However, they can also be used
to retrieve gene-specific annotation on the fly by specifying the
annotFun parameter, e.g. using the
annotateEntrez or annotateEnsembl functions,
provided in iSEE.
Alternatively, users can create a customized annotation function; for
more details on this, please consult the manual pages
?annotateEntrez and ?annotateEnsembl.
Column data tables
A Column data table contains the values of the
colData slot for the
SingleCellExperiment/SummarizedExperiment
object. Its behavior mirrors the implementation for the Row data
table. Correspondingly, if none are available, a column named
Present is added and set to TRUE for all
samples, to avoid issues with DT::datatable and an empty
DataFrame. Typically, these tables are used to link to
other plots to determine the samples to use for plotting or
coloring.
Heat maps
Heat map panels provide a compact overview of the data for
multiple features in the form of color-coded matrices. These correspond
to the assays stored in the
SummarizedExperiment object, where features (e.g., genes)
are the rows and samples are the columns.
User can select features (rows) to display from the selectize widget (which supports autocompletion), or also via other panels, like row data plots or row data tables. In addition, users can rapidly import custom lists of feature names using a modal popup that provides an Ace editor where they can directly type of paste feature names, and a file upload button that accepts text files containing one feature name per line. Users should remember to click the “Apply” button before closing the modal, to update the heat map with the new list of features.
The “Suggest feature order” button clusters the rows, and also
rearranges the elements in the selectize according to the clustering. It
is also possible to choose which assay type is displayed
("logcounts" being the default choice, if available).
Samples in the heat map can also be annotated, simply by selecting
relevant column metadata. A zooming functionality is also available,
restricted to the y-axis (i.e., allowing closer inspection on the
individual features included).
Description of iSEE functionality
Coloring plots by sample attributes
Column-based plots are the reduced dimension, feature assay and column data plots, where each data point represents a sample. Here, data points can be colored in different ways:
- The default is no color scheme (
"None"in the radio button). This results in data points of a constant user-specified color. - Any column of
colData(se)can be used. The plot automatically adjusts the scale to use based on whether the chosen column is continuous or categorical. - The assay values of a particular feature in each sample can be used.
The feature can be chosen either via a linked row table or selectize
input (as described for the Feature assay plot panel). Users
can also specify the
assaysfrom which values are extracted. - The identity of a particular sample can be used, which will be highlighted on the plot in a user-specified color. The sample can be chosen either via a linked column table or via a selectize input.
For row-based plots (i.e., the sample assay and row data plots), each data point represents a feature. Like the column-based plots, data points can be colored by:
-
"None", yielding data points of fixed color. - Any column of
rowData(se). - The identity of a particular feature, which is highlighted in the user-specified color.
- Assay values for a particular sample.
Fine control of the color maps is possible through the
ExperimentColorMap class, see this
vignette for more details.
Controlling point aesthetics
Data points can be set to different shapes according to categorical
factors in colData(se) (for column-based plots) or
rowData(se) (for row-based plots). This is achieved by
checking the "Shape" box to reveal the shape-setting
options. The size and opacity of the data points can be modified via the
options available by checking the "Point" box. This may be
useful for aesthetically pleasing visualizations when the number of
points is very large or small.
Faceting
Each point-based plot can be split into multiple facets using the
options in the "Facet" checkbox. Users can facet by row
and/or column, using categorical factors in colData(se)
(for column-based plots) or rowData(se) (for row-based
plots). This provides a convenient way to stratify points in a single
plot by multiple factors of interest. Note that point selection can only
occur within a single facet at a time; points cannot be
selected across facets.
FAQ
Q: Can you implement a ‘Copy to clipboard’ button in the code editor?
A: This is not necessary, as one can click anywhere in the code editor and instantly select all the code using a keyboard shortcut that depends on your operating system.
Q: When brushing with a transparency effect, it seems that data points in the receiving plot are not made transparent/subsetted correctly.
A: What you see is an artefact of overplotting: in areas excessively dense in points, transparency ceases to be an effective visual effect.
Q: Brushing on violin or square plots doesn’t seem to select anything.
A: For violin plots, points will be selected only if the brushed area includes the center of the x-tick, i.e., the center of the violin plot. This is intentional as it allows easy selection of all points in complex grouped violin plots. Indeed, the location of a specific point on the x-axis has no meaning. The same logic applies to the square plots, where only the center of each square needs to be selected to obtain all the points in the square.
Q: I’d like to try iSEE but I can’t install it/I just want a quick peek. Is there something you can do?
A: We set up an instance of iSEE running on the allen
dataset at this address: http://shiny.imbei.uni-mainz.de:3838/iSEE. A range of
interactive tours showcasing a variety of data types is also available
here: https://github.com/iSEE/iSEE2018. Please keep in mind
this is only for demonstration purposes, yet those instances show how
you or your system administrator can setup iSEE for
analyzing and/or sharing your
SummarizedExperiment/SingleCellExperiment
precomputed object.
Q: I would like to use iSEE with my
Seurat object, how do I do?
A: The Seurat package
provides as.SingleCellExperiment() to coerce
Seurat objects to SingleCellExperiment
objects. This conversion includes assays, cell metadata, feature
metadata, and dimensionality reduction results. You can then use the
SingleCellExperiment object as usual.
Additional information
Bug reports can be posted on the Bioconductor support site or raised as issues in the iSEE GitHub repository. The GitHub repository also contains the development version of the package, where new functionality is added over time. The authors appreciate well-considered suggestions for improvements or new features, or even better, pull requests.
If you use iSEE for your analysis, please cite it as shown below:
citation("iSEE")
#> To cite package 'iSEE' in publications use:
#>
#> Rue-Albrecht K, Marini F, Soneson C, Lun ATL (2018). "iSEE:
#> Interactive SummarizedExperiment Explorer." _F1000Research_, *7*,
#> 741. doi:10.12688/f1000research.14966.1
#> <https://doi.org/10.12688/f1000research.14966.1>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Article{,
#> title = {iSEE: Interactive SummarizedExperiment Explorer},
#> author = {Kevin Rue-Albrecht and Federico Marini and Charlotte Soneson and Aaron T. L. Lun},
#> publisher = {F1000 Research, Ltd.},
#> journal = {F1000Research},
#> year = {2018},
#> month = {Jun},
#> volume = {7},
#> pages = {741},
#> doi = {10.12688/f1000research.14966.1},
#> }Session Info
sessionInfo()
#> R version 4.6.1 (2026-06-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] scater_1.40.2 ggplot2_4.0.3
#> [3] scuttle_1.22.0 scrapper_1.6.3
#> [5] scRNAseq_2.26.0 iSEE_2.25.1
#> [7] SingleCellExperiment_1.34.0 SummarizedExperiment_1.42.0
#> [9] Biobase_2.72.0 GenomicRanges_1.64.0
#> [11] Seqinfo_1.2.0 IRanges_2.46.0
#> [13] S4Vectors_0.50.1 BiocGenerics_0.58.1
#> [15] generics_0.1.4 MatrixGenerics_1.24.0
#> [17] matrixStats_1.5.0 BiocStyle_2.40.0
#>
#> loaded via a namespace (and not attached):
#> [1] splines_4.6.1 later_1.4.8 BiocIO_1.22.0
#> [4] bitops_1.0-9 filelock_1.0.3 tibble_3.3.1
#> [7] XML_3.99-0.23 lifecycle_1.0.5 httr2_1.2.3
#> [10] doParallel_1.0.17 lattice_0.22-9 ensembldb_2.36.1
#> [13] alabaster.base_1.12.1 magrittr_2.0.5 sass_0.4.10
#> [16] rmarkdown_2.31 jquerylib_0.1.4 yaml_2.3.12
#> [19] httpuv_1.6.17 otel_0.2.0 DBI_1.3.0
#> [22] RColorBrewer_1.1-3 abind_1.4-8 Rtsne_0.17
#> [25] AnnotationFilter_1.36.0 RCurl_1.98-1.19 rappdirs_0.3.4
#> [28] circlize_0.4.18 ggrepel_0.9.8 irlba_2.3.7
#> [31] alabaster.sce_1.12.0 pkgdown_2.2.1 codetools_0.2-20
#> [34] DelayedArray_0.38.2 DT_0.34.0 tidyselect_1.2.1
#> [37] shape_1.4.6.1 UCSC.utils_1.8.0 farver_2.1.2
#> [40] viridis_0.6.5 ScaledMatrix_1.20.0 shinyWidgets_0.9.1
#> [43] BiocFileCache_3.2.0 GenomicAlignments_1.48.0 jsonlite_2.0.0
#> [46] GetoptLong_1.1.1 BiocNeighbors_2.6.0 iterators_1.0.14
#> [49] systemfonts_1.3.2 foreach_1.5.2 tools_4.6.1
#> [52] ragg_1.5.2 Rcpp_1.1.2 glue_1.8.1
#> [55] gridExtra_2.3.1 SparseArray_1.12.2 xfun_0.60
#> [58] mgcv_1.9-4 GenomeInfoDb_1.48.0 dplyr_1.2.1
#> [61] HDF5Array_1.40.0 gypsum_1.8.0 shinydashboard_0.7.3
#> [64] withr_3.0.3 BiocManager_1.30.27 fastmap_1.2.0
#> [67] rhdf5filters_1.24.0 shinyjs_2.1.1 rsvd_1.0.5
#> [70] digest_0.6.39 R6_2.6.1 mime_0.13
#> [73] textshaping_1.0.5 colorspace_2.1-3 listviewer_4.0.0
#> [76] RSQLite_3.53.3 cigarillo_1.2.0 h5mread_1.4.0
#> [79] rtracklayer_1.72.0 httr_1.4.8 htmlwidgets_1.6.4
#> [82] S4Arrays_1.12.0 pkgconfig_2.0.3 gtable_0.3.6
#> [85] blob_1.3.0 ComplexHeatmap_2.28.0 S7_0.2.2
#> [88] XVector_0.52.0 htmltools_0.5.9 bookdown_0.47
#> [91] ProtGenerics_1.44.0 rintrojs_0.3.4 clue_0.3-68
#> [94] scales_1.4.0 alabaster.matrix_1.12.0 png_0.1-9
#> [97] knitr_1.51 rjson_0.2.23 nlme_3.1-169
#> [100] curl_7.1.0 shinyAce_0.4.4 cachem_1.1.0
#> [103] rhdf5_2.56.0 GlobalOptions_0.1.4 BiocVersion_3.23.1
#> [106] parallel_4.6.1 miniUI_0.1.2 vipor_0.4.7
#> [109] AnnotationDbi_1.74.0 restfulr_0.0.17 desc_1.4.3
#> [112] pillar_1.11.1 grid_4.6.1 alabaster.schemas_1.12.0
#> [115] vctrs_0.7.3 promises_1.5.0 BiocSingular_1.28.0
#> [118] dbplyr_2.6.0 beachmat_2.28.0 xtable_1.8-8
#> [121] cluster_2.1.8.2 beeswarm_0.4.0 evaluate_1.0.5
#> [124] GenomicFeatures_1.64.0 cli_3.6.6 compiler_4.6.1
#> [127] Rsamtools_2.28.0 rlang_1.3.0 crayon_1.5.3
#> [130] ggbeeswarm_0.7.3 fs_2.1.0 viridisLite_0.4.3
#> [133] alabaster.se_1.12.0 BiocParallel_1.46.0 Biostrings_2.80.1
#> [136] lazyeval_0.2.3 colourpicker_1.3.0 Matrix_1.7-5
#> [139] ExperimentHub_3.2.0 bit64_4.8.2 Rhdf5lib_2.0.0
#> [142] KEGGREST_1.52.2 shiny_1.14.0 alabaster.ranges_1.12.0
#> [145] AnnotationHub_4.2.2 fontawesome_0.5.3 igraph_2.3.3
#> [148] memoise_2.0.1 bslib_0.11.0 bit_4.6.0